Questões Militares
Sobre validação de métodos analíticos e garantia da qualidade em farmácia
Foram encontradas 149 questões
I. Erros sistemáticos: têm sempre a mesma direção e são previsíveis. Suas fontes podem ser o instrumento, a calibração ou o operador. Por serem previsíveis e de fonte conhecida, é relativamente simples alcançar sua eliminação completa.
II. Erros aleatórios: podem ser negativos ou positivos, com direção e magnitudes imprevisíveis. Ocorrem sempre, mas não é possível determinar o seu valor. Não são elimináveis; porém, podem ser minimizados.
III. Monitoramento da precisão dos resultados: é feito através do controle intralaboratorial, que consiste na análise diária de amostras-controle, com valores conhecidos, dosadas, simultaneamente, com as amostras dos pacientes.
Está correto o que se afirma apenas em
Avalie se, na validação de métodos bioanalíticos para o laboratório de análise clínicas, são necessários os seguintes métodos de validação:
I. Precisão, exatidão e linearidade.
II. Limite inferior de quantificação, de detecção e robustez.
III. Carry-over, instabilidade e análise de efeito matriz.
Está correto o que se afirma em
Segundo GIL, E. S. (2010), uma forma de garantir a qualidade dos medicamentos fabricados é a utilização de ensaios de doseamento dos medicamentos. Esses métodos visam quantificar o teor de substância ativa em medicamentos e podem ser divididos em métodos clássicos ou instrumentais. Informe se é verdadeiro ou falso e a seguir marque a opção com a sequência correta:
(___) Para o doseamento de qualquer tipo de matéria ou produto manufaturado é necessário uma solução padrão. Essa solução deve ser preparada com um padrão primário em que o total de impurezas não deve exceder 0,1 a 0,2%.
(___) Na volumetria de complexação, a reação baseia-se em reações que envolvem um íon metálico e um agente ligante com formação de um complexo suficientemente estável. Os complexos formados com o ácido etilenodiaminotetracético (EDTA) são os mais comuns.
(___) A espectrometria de absorção no UV-visível é um dos mais uteis instrumentos de medida, para isso é necessário à obtenção de uma curva analítica ideal (45º em relação ao ponto de origem). Curvas com ângulos superiores a 45° (muito obtusos) são pouco sensíveis.
(___) A cromatografia gasosa (CG) está presente como método de escolha para doseamento de fármacos nas monografias da Farmacopeia Americana 24 ed., embora apresente ótima resolução e sensibilidade, ela não se aplica a produtos de baixa estabilidade térmica.
Segundo o Guia de dissolução aplicável a medicamentos genéricos, novos e similares (Guia nº 14 – versão 1 – ANVISA -2018), A Dissolução é o processo de liberação do insumo farmacêutico ativo (IFA) de sua forma farmacêutica, tornando-o disponível para absorção. O ensaio de dissolução é um teste físico-químico importante para demonstrar in vitro o desempenho de produtos que necessitam de dissolução para absorção e, consequente, efeito terapêutico.
O volume do meio de dissolução empregado depende, em grande parte, da solubilidade do IFA e da capacidade em manter a condição sink, cujo alcance é importante para evitar que a velocidade de dissolução seja influenciada, de maneira artificial, pela aproximação da saturação durante a realização do teste.
A condição sink é definida como sendo no mínimo três vezes o volume de meio necessário para se obter uma solução saturada do IFA, considerando a maior dose comercializada do produto. Nesta etapa, utiliza-se a maior dose comercializada, pois o ensaio de dissolução será realizado com apenas uma unidade farmacotécnica.
Avalie a seguinte situação:
Um farmacêutico deseja determinar a condição sink de um determinado IFA em diferentes meios de dissolução. A solubilidade desse IFA foi verificada em diferentes pH, e os resultados estão apresentados na Tabela 1.
Ele utilizará 500 ml de meio para realizar os testes de dissolução, considerando um comprimido contendo 8 mg de IFA.
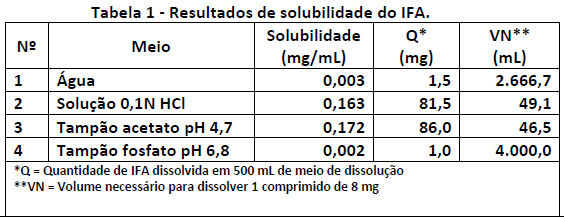
De acordo com os resultados acima, conclui-se que a condição sink será alcançada:
Os medicamentos registrados e comercializados no Brasil devem ser rotulados de acordo com recomendações específicas, visando garantir a informação correta promovendo segurança do uso. Os rótulos das embalagens primárias e secundárias dos medicamentos devem conter o nome comercial, a concentração dos princípios ativos e a via de administração.
A esse respeito, é correto afirmar que os princípios ativos devem ser escritos em letras minúsculas, utilizando a
Para registro de medicamento genérico, é necessário que a empresa apresente à Agência Nacional de Vigilância Sanitária (ANVISA) o estudo de Equivalência Farmacêutica.
A esse respeito, avalie alguns critérios para a realização deste estudo.
I. O estudo deverá comparar, simultaneamente, Medicamento Teste e Medicamento de Referência.
II. O estudo deverá ser realizado com lotes do Medicamento Teste e Medicamento de Referência dentro do prazo de validade.
III. O estudo deverá ser realizado por Centro de Equivalência Farmacêutica devidamente habilitado pela Anvisa para essa finalidade.
IV. O estudo deverá ser realizado após à conclusão do Estudo de Biodisponibilidade Relativa/ Bioequivalência, quando aplicável à forma farmacêutica.
Está correto apenas o que se afirma em
O desenvolvimento de medicamentos genéricos no Brasil deve responder a diversos quesitos que asseguram sua eficácia, segurança e intercambialidade com o medicamento de referência. Diversos parâmetros devem ser avaliados, como a biodisponibilidade, bioequivalência, equivalência farmacêutica e perfil de dissolução comparativo.
A esse respeito, analise as asserções a seguir e a relação proposta entre elas.
I . Biodisponibilidade à comparação da forma farmacêutica, via de administração e concentração do princípio ativo entre o produto de referência e genérico, indicando apresentarem a mesma farmacocinética
PORQUE
II . a equivalência farmacêutica refere-se à comprovação de que o medicamento de referência e genérico apresentam resultados in vitro similares, indicando apresentarem a mesma eficácia e segurança.
Sobre essas asserções, é correto afirmar que
Os sistemas de tratamento de ar têm um papel importante na qualidade de produtos farmacêuticos. Além de oferecerem proteção ao produto durante etapas de produção, também fornecem condições confortáveis e seguras aos operadores e os protegem de contaminantes provenientes do processo fabril.
Avalie os parâmetros que devem ser monitorados a fim de atender aos requisitos de qualidade de ar em áreas produtivas.
I. Umidificação por atomização.
II. Filtragem.
III. Desumidificação.
IV. Ventilação.
Está correto apenas o que se afirma em
Teste de estabilidade de formulações farmacêuticas é um conjunto de ensaios que visa definir o prazo de validade, as condições de armazenamento do produto e o material de embalagem que deve ser utilizado para a formulação.
Considerando os estudos de estabilidade que devem ser realizados nas formulações farmacêuticas, analise as asserções a seguir e a relação proposta entre elas.
I . O teste de estabilidade acelerado submete as amostras a condições forçadas de armazenamento, com valores de temperatura acima do recomendado para o armazenamento da formulação
PORQUE
II . condições elevadas de temperatura e umidade promovem estresse na formulação, acelerando reações de decomposição, que podem predizer a estabilidade da formulação ao longo do tempo.
Sobre essas asserções, é correto afirmar que
De acordo com a Instrução Normativa Nº 2, de 30 de março de 2009, da Agência Nacional de Vigilância Sanitária, lotes piloto são aqueles lotes de produtos farmacêuticos produzidos por um processo representativo e reprodutivo de um lote de produção em escala industrial, sendo os mesmos essenciais para a avaliação criteriosa quanto às características e à qualidade de um produto.
A característica correta necessária ao lote piloto para que este seja designado como tal, é a de que ele
As embalagens utilizadas na indústria farmacêutica servem para acondicionar, proteger os produtos contra degradações e transmitir informações ao consumidor.
A esse respeito é correto afirmar que está intimamente em contato com a formulação farmacêutica, evitando reações químicas ou biológicas, a embalagem
Em relação aos medicamentos genéricos e similares, que podem ser dispensados dos estudos de biodisponibilidade relativa/bioequivalência, analise as asserções a seguir e a relação proposta entre elas.
I. A bioisenção, baseada no sistema de classificação biofarmacêutica, pode ser aplicada a medicamentos genéricos e similares, a medicamentos novos, de liberação imediata, quando administrados por via oral e formulados com excipientes que não apresentem impacto sobre a biodisponibilidade
PORQUE
II. o fármaco é altamente solúvel, solubiliza-se completamente em até 250ml em soluções tampão utilizadas dentro da faixa de pH fisiológico (1,2 a 6,8), a 37 ± 1ºC.
Sobre essas asserções, é correto afirmar que