Questões de Concurso
Sobre hematologia em medicina
Foram encontradas 4.528 questões
O hemograma revela: Hb 8,7 g/dL | Ht 26% | VCM 68 fL | CHCM 28 g/dL | RDW 18% | Leucócitos e plaquetas normais. Ferritina: 10 ng/mL. Ferro sérico: 25mcg/dL, TIBIC: 420mcg/dL, saturação de transferrina: 6%, o PCR está abaixo do limite de detecção.
A principal suspeita diagnóstica é de
Paciente masculino, 55 anos, procurou o consultório médico com queixas de fadiga progressiva, fraqueza, sangramento gengival esporádico e surgimento fácil de equimoses nos últimos 3 meses. Negou febre, calafrios, perda ponderal significativa ou sudorese noturna. Não havia outras comorbidades conhecidas. O paciente não estava em uso recente de medicamentos que pudessem justificar o quadro. Ele relatou histórico de consumo moderado de álcool (cerca de 2 a 3 doses por dia), há aproximadamente 20 anos.
Ao exame físico, apresentava-se pálido, com algumas petéquias discretas em membros inferiores. Não havia linfonodomegalias palpáveis. O fígado era palpável há 2 cm abaixo do rebordo costal direito, de consistência normal e indolor. Baço não foi palpável. O restante do exame físico estava sem alterações significativas.
Exames laboratoriais iniciais - Hemograma:
Hemoglobina: 8,0 g/dL; Leucócitos totais: 2.500/mm³ com Neutrófilos: 1.000/mm³; Plaquetas: 70.000/mm³. Volume Corpuscular Médio (VCM): 105 fL; Anisocitose (+); Plaquetas hipogranulares. Reticulócitos: 0,5%. Cinética de Ferro: Ferro sérico: 90 mcg/dL (VR: 60-170 mcg/dL); Ferritina: 150 ng/mL (VR: 20-250 ng/mL); Capacidade Total de Ligação do Ferro (TIBC): 300 mcg/dL (VR: 250-400 mcg/dL). Bioquímica: Bilirrubina total: 1,5 mg/dL com bilirrubina indireta: 1,0 mg/dL; AST: 60 U/L; ALT: 75 U/L; Gama-GT (GGT): 120 U/L (VR: <50 U/L); Creatinina: 1,0 mg/dL.
Considerando o quadro clínico e os exames laboratoriais iniciais, a abordagem investigativa mais apropriada para determinar a etiologia da pancitopenia é
Paciente feminina, 28 anos, foi internada pela quarta vez em um hospital de referência. As internações anteriores, ocorridas aos 13, 18 e 26 anos, estiveram relacionadas a episódios de sangramento atípico. Aos 13 anos, sangrou prolongadamente após uma amigdalectomia, necessitando de transfusão sanguínea, sendo a investigação inicial inconclusiva. Aos 18, apresentou sangramento uterino disfuncional que exigiu hospitalização, sem identificação de distúrbio da coagulação nem plaquetopenia. Aos 26, teve esquimoses extensas em ambos os braços após uma leve contusão. Atualmente, a paciente procurou o pronto-socorro com fraqueza progressiva e tontura. Havia realizado uma biópsia de pele recente para remoção de nevo suspeito, mas o sangramento do local persistiu por mais de 48 horas, motivando o uso de analgésicos e anti-inflamatórios não esteroidais.
Ao exame físico, a paciente estava pálida, hipocorada, com frequência cardíaca de 105 bpm e pressão arterial de 105 x 60 mmHg. Estava lúcida, orientada, eutrófica e não apresentava sinais de gravidade aguda ou infecção. Negava uso de medicação regular ou doença prévia, exceto pelos episódios de sangramento. Seus exames demonstraram uma piora gradual da anemia: Hemoglobina 10 g/dL aos 26 anos, e atualmente 7,5 g/dL. Durante a internação, relatou alguns episódios de enterorragia discreta. A investigação de anemia demonstrou morfologia microcítica e hipocrômica, com reticulócitos corrigidos de 1,2%, ferritina 10 ng/mL, índice de saturação de transferrina 9%, TIBC: 420 mcg/dL. O médico responsável iniciou a investigação de anemia com colonoscopia, que revelou divertículo de Meckel, com erosões superficiais sem sangramento ativo. Ao revisar os exames, observou plaquetas de 160.000/mm³ e um coagulograma normal (TP e PTTa dentro dos limites).
Considerando o histórico clínico, os achados do exame físico e os resultados laboratoriais, a conduta mais apropriada no momento seria
Nos últimos 10 dias, evoluiu com quadro de linfonodomegalia generalizada, hepatoesplenomegalia, derrame pleural, febre, mal estar, hipotensão, taquicardia, sudorese, configurando uma forma agressiva e de rápida evolução. Exames complementares apresentando linfocitose acentuada, elevação da lactato desidrogenase 9x o normal, aumento do cálcio sérico e leões cutâneas.
O técnico de laboratório contactou o médico assistente pois no sangue periférico haviam as células atípicas exibidas abaixo, e que definiram o diagnóstico da enfermidade da paciente.
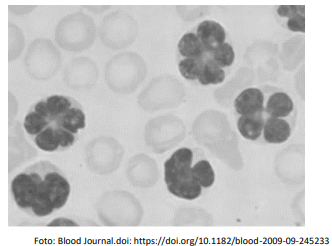
O diagnóstico da doença em questão e o agente etiológico são, respectivamente,
A principal hipótese diagnóstica é leucemia
Paciente feminina, 62 anos, procura atendimento por fadiga progressiva há 6 meses, associada a episódios de confusão mental, irritabilidade e dificuldade de concentração. Refere ainda parestesias em membros inferiores com sensação de “queimação” nos pés e instabilidade à marcha. Nega etilismo, tabagismo ou uso de medicamentos.
Ao exame físico: palidez cutâneo-mucosa (2+/4+), glossite atrófica, diminuição da sensibilidade vibratória e posicional em membros inferiores, sinal de Romberg positivo. Exames laboratoriais: Hb 8,2 g/dL, VCM 110 fL, leucócitos 3.200/mm³, plaquetas 98.000/mm³, LDH 890 U/L (VR: 120-480), bilirrubina indireta 2,8 mg/dL, ácido fólico normal. Esfregaço de sangue periférico revela neutrófilos hipersegmentados e macro-ovalócitos. Para estabelecer o diagnóstico etiológico mais provável, a combinação de exames mais adequada é
Paciente do sexo masculino, de 68 anos, foi recentemente diagnosticado com leucemia mieloide aguda. Após 7 dias do início do tratamento quimioterápico apresenta importante elevação das escórias nitrogenadas associada a hiperuricemia, hipocalcemia e hiperfosfatemia. Apesar dos cuidados clínicos, evoluiu para Injúria Renal Aguda (IRA), estágio 3, vindo a necessitar de terapia renal substitutiva.
Nesse caso, a hipótese diagnóstica é
Uma mulher de 63 anos, portadora de plaquetopenia crônica, com contagem sempre em torno de 45 mil plaquetas por microlitro, sem episódios de sangramentos no dia a dia, foi submetida a um procedimento médico invasivo em que a transfusão de plaquetas não é indicada.
O procedimento em questão é a
Uma paciente de 29 anos é admitida no CTI após ser submetida auma esplenectomia laparoscópica sem complicações, por umapúrpura trombocitopênica idiopática.
No segundo dia de pós-operatório, ela se recupera bem, estáafebril, hemodinamicamente estável e sem queixas. Seuhemograma revela uma contagem sérica de plaquetas de654.000/μL.
A causa mais provável de trombocitose nessa paciente é
Diante dessa situação, o melhor exame complementar para confirmar ou afastar o diagnóstico de TVP é
Pré-escolar, masculino, 2 anos e meio, previamente hígido, apresenta palidez, fraqueza, dor abdominal e icterícia 48 horas após iniciar tratamento para infecção do trato urinário com nitrofurantoína.
Hemograma evidencia anemia normocítica e normocrômica. Os reticulócitos e a desidrogenase láctica estão elevados.
A hipótese diagnóstica, nesse caso, é
Clinicamente, essas falências resultam em uma medula óssea hipoplástica e, no sangue periférico, em anemia, neutropenia ou trombocitopenia.
As condições clínicas mais frequentes de falências medulares são: Anemia Aplástica (AA), Aplasia Pura de Série Vermelha (APSV), Neutropenia Congênita Grave.
Conforme os PCDT do Ministério da Saúde, assinale a opção que apresenta um critério de inclusão para tratamento da falência medular.
Essas células deformadas ficam enrijecidas, não conseguem circular pelos pequenos vasos sanguíneos e se rompem com facilidade, o que causa bloqueios no fluxo sanguíneo, dor intensa (crises de dor), anemia, lesões em órgãos e maior suscetibilidade a infecções.
Assinale a opção que apresenta um critério de exclusão para tratamento da doença falciforme com hidroxiureia.
A presença de trombofilias adquiridas e hereditárias aumentam o risco de TEV na gravidez.
A respeito da trombofilia, avalie as afirmativas a seguir.
I. A trombofilia adquirida mais relevante é a Síndrome Antifosfolipídeo (SAF), que pode cursar com manifestações venosas e arteriais.
II. As formas de trombofilia hereditária, em ordem de relevância na gravidez, são: mutações genéticas no fator V de Leiden; mutação no gene da protrombina; deficiências de antitrombina, de proteína C e de proteína S.
III. As trombofilias hereditárias têm maior relação com manifestações arteriais.
Está correto o que se afirma em
Ao exame físico, apresentava palidez cutâneo-mucosa 2+/4+, frequência cardíaca de 102 bpm, pressão arterial de 110 x 70 mmHg e sopro sistólico 2+/6+ no foco mitral. O restante do exame físico não apresentava alterações. Foram solicitados exames complementares para investigação diagnóstica.
Os parâmetros laboratoriais que caracterizam adequadamente o diagnóstico de anemia ferropriva, nessa paciente, compreendem
Ao exame físico, apresenta palidez cutaneomucosa, glossite atrófica e discreta icterícia escleral. Não há sinais de sangramento ativo.
Os exames laboratoriais revelam:
• Hemoglobina: 8,2 g/dL (VR: 12-16 g/dL);
• Hematócrito: 25% (VR: 36-48%);
• VCM: 118 fL (VR: 80-100 fL);
• CHCM: 33 g/dL (VR: 32-36 g/dL);
• Contagem de Reticulócitos: 0,3% (VR: 0,5-2,5% ou 25.000- 75.000/mm³);
• Leucócitos totais: 3.200/mm³ (VR: 4.000-10.000/mm³);
• Neutrófilos: 45% (VR: 40-70%);
• Plaquetas: 90.000/mm³ (VR: 150.000-450.000/mm³);
• Bilirrubina total: 2,5 mg/dL (VR: 0,2-1,2 mg/dL);
• Bilirrubina indireta: 2,1 mg/dL (VR: < 0,8 mg/dL);
• LDH: 980 U/L (VR: 120-240 U/L);
• Vitamina B12 sérica: 80 pg/mL (VR: 200-900 pg/mL);
• Folato sérico: 12 ng/mL (VR: 2,7-17 ng/mL);
• Ácido Metilmalônico (AMM) urinário: elevado; e
• Homocisteína plasmática: elevada.
Sobre o caso, analise as afirmativas a seguir.
I. A vitamina B12 e o folato atuam como cofatores na síntese de RNA, e sua deficiência compromete a divisão celular, levando à produção de células pequenas e hipocrômicas, com a deficiência de folato sendo a principal causa de sintomas neurológicos devido ao acúmulo de homocisteína, que é neurotóxica.
II. A deficiência de vitamina B12 e de folato afeta primariamente a síntese de proteínas estruturais das membranas celulares, resultando em eritrócitos frágeis e suscetíveis à hemólise intravascular, com a medula óssea respondendo com eritropoiese compensatória, e os sintomas neurológicos são decorrentes da isquemia cerebral crônica.
III. A vitamina B12 e o folato são essenciais para a síntese de DNA, e sua deficiência causa maturação citoplasmática assíncrona, resultando em eritrócitos grandes e imaturos, além de afetar outras linhagens celulares; a deficiência de B12 leva ao acúmulo de AMM e homocisteína, contribuindo para neuropatia.
Está correto o que se afirma em
Entre essas afecções encontramos a Doença de Alexander, que é causada por deficiência do fator