Questões de Concurso
Sobre validação de métodos analíticos e garantia da qualidade em farmácia
Foram encontradas 578 questões
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Farmacocinética |
Q569123
Farmácia
Determinar a estabilidade do fármaco em matrizes biológicas
é de extrema importância. Para a realização do estudo de
estabilidade devem ser observados os parâmetros de
exatidão, precisão, linearidade, limite de detecção, limite de
quantificação, especificidade, limite de variação e robustez,
previamente validados. Não faz parte dos ensaios de
estabilidade:
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Farmacocinética |
Q569122
Farmácia
Na validação de um método bioanalítico é necessária a
construção das corridas analíticas que contém as amostras
de branco, a curva de calibração e as amostras controle de
qualidade. Na execução de uma validação para fins de
estudos de bioequivalência, o valor da concentração da
amostra de controle de qualidade baixa deve ser:
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Farmacocinética |
Q569121
Farmácia
A especificidade é a habilidade do método bioanalítico de
medir e diferenciar o analito de componentes que possam
estar presentes na amostra, tais como metabólitos,
impurezas, compostos de degradação ou componentes da
matriz. Na verificação da especificidade:
I. caso o método seja destinado à quantificação de mais de um fármaco, todos devem ser injetados para determinar os tempos de retenção.
II. deve-se analisar amostras de branco da matriz biológica obtidas de oito indivíduos, sendo seis amostras normais, uma lipêmica e uma hemolisada.
III. os resultados das amostras de branco devem ser comparados com aqueles obtidos com solução aquosa do analito, em concentração próxima ao limite superior de quantificação.
IV. os interferentes podem ser componentes da matriz biológica, metabólitos, produtos de decomposição e medicamentos utilizados concomitantemente ao estudo.
Assinale:
I. caso o método seja destinado à quantificação de mais de um fármaco, todos devem ser injetados para determinar os tempos de retenção.
II. deve-se analisar amostras de branco da matriz biológica obtidas de oito indivíduos, sendo seis amostras normais, uma lipêmica e uma hemolisada.
III. os resultados das amostras de branco devem ser comparados com aqueles obtidos com solução aquosa do analito, em concentração próxima ao limite superior de quantificação.
IV. os interferentes podem ser componentes da matriz biológica, metabólitos, produtos de decomposição e medicamentos utilizados concomitantemente ao estudo.
Assinale:
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Farmacocinética |
Q569120
Farmácia
Um método bioanalítico desenvolvido em um laboratório só
poderá ser aplicado a um estudo de bioequivalência/
biodisponibilidade após a etapa de validação. Validações
parciais devem ser realizadas quando ocorrerem
modificações no método bioanalítico já validado. As
mudanças típicas que podem requerer uma validação parcial
não incluem:
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Farmacocinética |
Q569119
Farmácia
Para os estudos de biodisponibilidade relativa/
bioequivalência deve-se utilizar padrão interno, sempre que
métodos cromatográficos forem utilizados. Quando seu uso
não for possível deve-se justificar. O padrão interno é um
composto, geralmente com características estruturais
similares ao analito:
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Farmacocinética |
Q569117
Farmácia
O delineamento de um estudo de bioequivalência do tipo
cruzado replicado é recomendado:
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Farmacocinética |
Q569113
Farmácia
A ANVISA estabeleceu o regulamento técnico para o registro
de medicamentos genéricos no ano de 1999. Segundo esta
regulamentação, revogada pela Resolução – RDC nº 10 de 2
janeiro de 2001, para se registrar um medicamento como
genérico, é necessário que se comprove sua equivalência
farmacêutica e bioequivalência em relação ao medicamento
de referência. Com relação às afirmativas sobre equivalência
farmacêutica, assinale a alternativa incorreta.
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Farmacocinética |
Q569103
Farmácia
Alguns fármacos apresentam grande afinidade às proteínas
plasmáticas. Um exemplo é o antiretroviral lopinavir que se
liga a alfa-glicoproteína e somente cerca de 1% do fármaco
permanece na forma livre. Em geral, o plasma sanguíneo é
utilizado como amostra para quantificar as concentrações do
fármaco e, desta forma, determinar a curva de concentração
versus o tempo para a análise de bioequivalência. Em casos
como o citado anteriormente, a estratégia de amostragem e
quantificação, segundo a legislação para estudos de
bioequivalência, deve ser:
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Farmacovigilância |
Q568687
Farmácia
No intuito de contribuir para o monitoramento da qualidade de medicamentos, a Agência Nacional de Vigilância Sanitária (ANVISA), em conjunto com o Instituto Nacional de Controle de Qualidade em Saúde, Fundação Oswaldo Cruz (INCQS/FIOCRUZ), instituiu o Programa Nacional de Verificação da Qualidade de Medicamentos (PROVEME). São critérios utilizados para a coleta de amostras para a verificação de qualidade:
I. medicamentos da rede SUS. II. medicamentos notificados com suspeitas de desvio da qualidade.
III. medicamentos de maior custo. IV. medicamentos analisados anteriormente que obtiveram resultado de análise insatisfatória.
Assinale:
Assinale:
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Flexografia |
Q568641
Farmácia
De acordo com RDC N°17, de 16 de abril de 2010, as Boas
Praticas de Fabricação de Medicamentos determinam que:
I. todos os processos de fabricação devam ser claramente definidos e sistematicamente revisados em função da experiência adquirida.
II. devem ser capazes de fabricar medicamentos dentro dos padrões de qualidade exigidos, atendendo às respectivas especificações.
III. sejam realizadas as qualificações e validações necessárias.
IV. que seja implantado um sistema capaz de recolher qualquer lote, após sua comercialização ou distribuição, na ocasião da identificação de algum desvio de qualidade. O prazo de recolhimento deverá ser de 45 a 60 dias.
Assinale:
I. todos os processos de fabricação devam ser claramente definidos e sistematicamente revisados em função da experiência adquirida.
II. devem ser capazes de fabricar medicamentos dentro dos padrões de qualidade exigidos, atendendo às respectivas especificações.
III. sejam realizadas as qualificações e validações necessárias.
IV. que seja implantado um sistema capaz de recolher qualquer lote, após sua comercialização ou distribuição, na ocasião da identificação de algum desvio de qualidade. O prazo de recolhimento deverá ser de 45 a 60 dias.
Assinale:
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Flexografia |
Q568639
Farmácia
A RDC N°17, de 16 de abril de 2010, considera que um
processo específico produzirá consistentemente um produto
que atenda suas especificações e atributos de qualidade.
Nesse artigo referente ao capitulo dedicado a qualificação e
validação, a norma em referência está se referindo a:
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Flexografia |
Q568637
Farmácia
Em consonância com a RDC N°17, de 16 de abril de 2010, os
estudos de validação são uma parte essencial das Boas
Praticas de Fabricação (BPF) e devem ser conduzidos de
acordo com:
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Flexografia |
Q568634
Farmácia
Com base na Resolução - RDC N° 17, de 16 de abril de 2010
que “Dispõe sobre as Boas Práticas de Fabricação de
Medicamentos", analise a tabela a seguir.
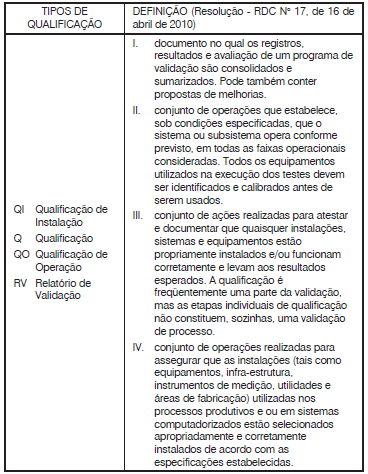
Está correto apenas o que se afirma em
Está correto apenas o que se afirma em
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Flexografia |
Q568631
Farmácia
A RDC N°17, de 16 de abril de 2010, preceitua que a
qualificação e a validação não devem ser consideradas
exercícios únicos. Após a aprovação do relatório de
qualificação e/ou validação deve haver um programa
contínuo de monitoramento, o qual deve ser embasado em
uma revisão periódica. O compromisso da manutenção da
situação de qualificação/validação deve estar descrito nos
documentos relevantes da empresa, como:
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Flexografia |
Q568629
Farmácia
De acordo RDC N°17, de 16 de abril de 2010, a extensão da
revalidação depende da natureza e da significância da
mudança. As mudanças não devem afetar adversamente a
qualidade do produto ou as características do processo. As
alterações de equipamentos que envolvam a substituição do
equipamento por um equivalente normalmente:
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Flexografia |
Q568626
Farmácia
Segundo a RDC N°17, de 16 de abril de 2010, a validação de
processos e sistemas é fundamental para se atingir os
objetivos. É por meio do projeto e validação que um
fabricante pode estabelecer com confiança que os produtos
fabricados irão consistentemente atender as suas
especificações.
A documentação associada à validação deve incluir:
I. Procedimentos Operacionais Padrão (POP).
II. especificações.
III. Plano Mestre de Validação (PMV).
IV. protocolos e relatórios de qualificação.
V. protocolos e relatórios de validação.
Assinale:
I. Procedimentos Operacionais Padrão (POP).
II. especificações.
III. Plano Mestre de Validação (PMV).
IV. protocolos e relatórios de qualificação.
V. protocolos e relatórios de validação.
Assinale:
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Flexografia |
Q568625
Farmácia
De acordo com o que estabelece a RDC N°17, de 16 de abril
de 2010, o fabricante é responsável pela qualidade dos
medicamentos por ele fabricados, assegurando que sejam
adequados aos fins a que se destinam, cumpram com os
requisitos estabelecidos em seu registro e não coloquem os
pacientes em risco por apresentarem segurança, qualidade
ou eficácia inadequada. O cumprimento deste objetivo é
responsabilidade:
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Flexografia |
Q568624
Farmácia
De acordo com a RDC N°17, de 16 de abril de 2010, são
exemplos de itens que devem ser verificados por ocasião da
aplicação do protocolo de Qualificação de Instalação (QI):
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Flexografia |
Q568621
Farmácia
Um aspecto essencial na validação de limpeza é determinar
quanto de limpeza é suficiente. Apesar de oficialmente não
endossar critérios adotados por indústrias farmacêuticas, o
FDA (Food Drug Administration) dos Estados Unidos da
América faz referência a critérios adotados pela empresa Eli
Lilly, que estabelece os seguintes critérios (LeBlanc, 1999):
I. O equipamento deve estar visualmente limpo.
II. Qualquer agente ativo do produto após a limpeza deve estar presente em níveis máximos de 10 ppm ou 10 mg/g do produto após a limpeza em relação ao produto subseqüente.
III. Qualquer agente ativo do produto após a limpeza deve estar presente em níveis máximos de 1/100 da dose mínima diária da substância ativa em relação à dose máxima diária do produto subseqüente, calculado de acordo com a equação seguinte (LeBlanc, 1999):
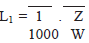
Onde:
L1 = Limite no produto subseqüente em mg/g
Z = Dose mínima diária do produto a ser limpo
Assinale:
I. O equipamento deve estar visualmente limpo.
II. Qualquer agente ativo do produto após a limpeza deve estar presente em níveis máximos de 10 ppm ou 10 mg/g do produto após a limpeza em relação ao produto subseqüente.
III. Qualquer agente ativo do produto após a limpeza deve estar presente em níveis máximos de 1/100 da dose mínima diária da substância ativa em relação à dose máxima diária do produto subseqüente, calculado de acordo com a equação seguinte (LeBlanc, 1999):
Onde:
L1 = Limite no produto subseqüente em mg/g
Z = Dose mínima diária do produto a ser limpo
Assinale:
Ano: 2010
Banca:
FGV
Órgão:
FIOCRUZ
Prova:
FGV - 2010 - FIOCRUZ - Tecnologista em Saúde - Flexografia |
Q568620
Farmácia
A Resolução - RDC N° 17, de 16 de abril de 2010 “Dispõe
sobre as Boas Práticas de Fabricação de Medicamentos"
preconiza que a requalificação deve ser realizada de acordo
com um cronograma definido. A freqüência de requalificação
pode ser determinada com base nos seguintes fatores: