Foram encontradas 4.871 questões
Resolva questões gratuitamente!
Junte-se a mais de 4 milhões de concurseiros!
I – Sendo o registro do produto feito como medicamento genérico, o QTPP deve ser elaborado considerando a comparação da formulação entre o produto teste e o produto de referência, baseada principalmente na composição dos excipientes, e não na comparação das características de desempenho.
II – O QTPP pode incluir características do produto em questão, como uso pretendido, via de administração e teor, assim como características de desempenho para os métodos analíticos utilizados, como precisão e exatidão.
III – O pH e a viscosidade podem ser considerados no QTPP do produto, podendo incluir no perfil alvo qualquer característica que será idealmente alcançada para garantir a qualidade do medicamento.
IV – Todas as características listadas no QTPP para o produto são definidas como os atributos críticos de qualidade (CQAs).
V – A elaboração do QTPP deve ser baseada nas características de qualidade do produto, favorecendo o desenvolvimento do medicamento com padrões de qualidade centrados no paciente.
Das afirmativas acima:
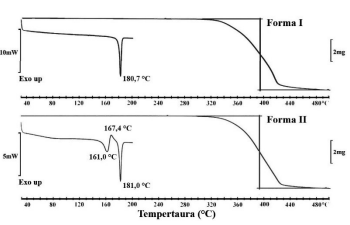
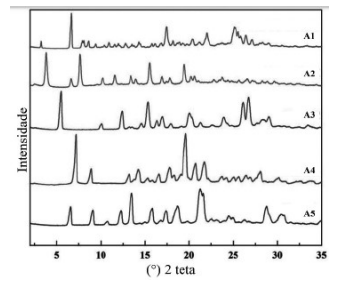
I – Os IFAs dos fabricantes A e B apresentam a mesma forma sólida.
II – Considerando a informação do DIFA que os IFAs são hidratos, os fabricantes B e C podem apresentar diferentes graus de hidratação ou são polimorfos do hidrato de mesma estequiometria.
III – A titulação Karl-Fisher permite a diferenciação da água de superfície e da água e incorporada na estrutura cristalina do hidrato, auxiliando na seleção da forma sólida para o desenvolvimento do produto.
IV – Considerando a informação do DIFA que os IFAs são hidratos, os fabricantes A, B e C podem ser selecionados para o desenvolvimento do produto, pois, já que as propriedades físico-químicas dos IFAs são as mesmas não haverá diferenças no desempenho da formulação.
V – É importante a avaliação da interconversão entre as formas sólidas do IFA, uma vez que, em suspensão, pode ocorrer a transição para a forma estável, resultando em crescimento cristalino e uma distribuição de tamanho de partícula indesejada.
VI – Nos casos de as diferenças nas formas sólidas apresentarem impacto no desempenho do produto, é importante monitorar possíveis interconversões durante o prazo de validade, por meio de técnicas capazes de discriminar formas cristalinas diferentes, tais como calorimetria diferencial exploratória, difração de raios X de pó e cromatografia líquida de alta eficiência.
Das afirmativas acima:
O teste de adequação do sistema (SST) é usado para verificar se o sistema de medição e as operações analíticas associadas ao procedimento analítico são adequados para a finalidade pretendida durante o período de análise e permitem a detecção de desempenho inaceitável. Observe as afirmativas a seguir em relação ao SST para métodos analíticos.
I – O SST não depende do tipo do procedimento analítico.
II – Os componentes do SST devem ser selecionados com base em avaliação de riscos bem como o conhecimento e a compreensão dos dados de desenvolvimento analítico.
III – O número de pratos teóricos, a relação sinal-ruído e o fator de simetria podem ser usados como parâmetros de SST para métodos por cromatografia líquida de alta eficiência.
IV – Nenhuma análise de amostra é aceitável, a menos que a adequação do sistema tenha sido demonstrada.
V – Para métodos de teor por cromatografia líquida de alta eficiência, quando um requisito de desvio padrão relativo é especificado igual ou inferior a 2,0%, o cálculo deve ser baseado em dados de três injeções replicadas do analito, conforme requisitos farmacopeicos.
Das afirmativas acima:
I – Não é permitido que a empresa solicite o registro de um medicamento em mais de um material de embalagem primária. Quando mais de uma embalagem primária for estudada no desenvolvimento, apenas a de maior proteção deve ser selecionada.
II – O vidro tipo I, também denominado vidro borossilicato, é o único tipo de vidro que pode ser utilizado para medicamentos de uso humano por ser totalmente inerte e não trazer riscos de incompatibilidade com a forma farmacêutica.
III – Polímeros termoplásticos podem ser usados para a tecnologia blow-fill-seal, na qual as etapas de formação da embalagem, enchimento com o produto farmacêutico e selagem do sistema final ocorrem em operação contínua.
IV – Embalagens termomoldáveis devem conter plastificantes em suas composições, permitindo a redução da temperatura de transição vítrea dos polímeros para que as temperaturas de processo possam ser reduzidas. São exemplos de plastificantes de embalagens termomoldáveis os ftalatos.
As afirmativas I, II e III e IV são respectivamente:
I – Meio dispersante.
II – Concentração da amostra.
III – Pureza química do padrão de referência.
IV – Faixa de obscuração.
Das afirmativas acima:
I – A utilização de água como meio de dissolução é recomendada especialmente para fármacos que se dissolvam de maneira dependente do pH do meio.
II - O volume do meio de dissolução empregado depende, em grande parte, da solubilidade do fármaco e da capacidade em manter a condição sink.
III - A determinação do poder discriminativo do método de dissolução pode ser realizada por meio da avaliação de lotes produzidos com alterações deliberadas dos atributos farmacêuticos.
As afirmativas I, II e III são respectivamente: