Questões de Concurso
Sobre controle de qualidade industrial em farmácia
Foram encontradas 1.680 questões
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Tecnologista em desenvolvimento analítico e de embalagem |
Q3341078
Farmácia
O programa de estabilidade de acompanhamento faz
parte das boas práticas de fabricação. Sobre o programa
de estabilidade, pode-se afirmar que:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Tecnologista em desenvolvimento analítico e de embalagem |
Q3341077
Farmácia
A RDC Nº 658 de 2022 dispõe sobre as diretrizes gerais
de boas práticas de fabricação de medicamentos e apresenta um capítulo específico para o controle de qualidade.
Sobre as análises realizadas em controle de qualidade,
pode-se afirmar que:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Tecnologista em desenvolvimento analítico e de embalagem |
Q3341069
Farmácia
Avalie se as afirmações abaixo são verdadeiras(V) ou
falsas(F) quanto aos estudos de fotoestabilidade:
I- A fotodegradação pode ocorrer por diversos mecanismos, alguns dos quais não são dependentes da quantidade de luz que o sistema é exposto. Assim, pode-se inferir que algumas reações de fotodegradação ocorrem mesmo se a exposição for a pequena quantidade de luz.
II- A luz pode atuar apenas como promotor de uma reação de degradação do ativo em um medicamento, que continua a ocorrer mesmo depois de cessada a exposição.
III- O estudo de fotoestabilidade, de acordo com o preconizado pela RDC 318 de 06 de novembro de 2019, baseia-se na exposição do produto a uma grande quantidade de luz em um curto período de tempo ao passo que na vida real do medicamento a exposição se dá a uma pequena quantidade de luz por um longo período de tempo.
IV- Em um estudo de fotoestabilidade o medicamento em desenvolvimento deve ser exposto tanto à incidência de luz visível (cuja unidade de luminosidade é o lux) quanto para a luz ultravioleta (cuja unidade de radiação é watt.horas/m2).
A ordem correta, de cima para baixo, é:
I- A fotodegradação pode ocorrer por diversos mecanismos, alguns dos quais não são dependentes da quantidade de luz que o sistema é exposto. Assim, pode-se inferir que algumas reações de fotodegradação ocorrem mesmo se a exposição for a pequena quantidade de luz.
II- A luz pode atuar apenas como promotor de uma reação de degradação do ativo em um medicamento, que continua a ocorrer mesmo depois de cessada a exposição.
III- O estudo de fotoestabilidade, de acordo com o preconizado pela RDC 318 de 06 de novembro de 2019, baseia-se na exposição do produto a uma grande quantidade de luz em um curto período de tempo ao passo que na vida real do medicamento a exposição se dá a uma pequena quantidade de luz por um longo período de tempo.
IV- Em um estudo de fotoestabilidade o medicamento em desenvolvimento deve ser exposto tanto à incidência de luz visível (cuja unidade de luminosidade é o lux) quanto para a luz ultravioleta (cuja unidade de radiação é watt.horas/m2).
A ordem correta, de cima para baixo, é:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Tecnologista em desenvolvimento analítico e de embalagem |
Q3341068
Farmácia
De acordo com o Guia número 28 de 11 de novembro
de 2019, o estudo de estabilidade de insumos farmacêuticos ativos e de medicamentos tem o objetivo de fornecer
evidências sobre como a qualidade dos produtos varia ao
longo do tempo, quando sob influência de diversos fatores
ambientais. Sobre os estudos de estabilidade de medicamentos, de acordo com a RDC 318 de 06 de novembro de
2019, é correto afirmar que:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Tecnologista em desenvolvimento analítico e de embalagem |
Q3341063
Farmácia
Avalie os resultados abaixo, conforme o preconizado pela RDC 318 de 6 de novembro de 2019, que estabelece os
critérios para a realização de estudos de estabilidade de insumos farmacêuticos ativos e medicamentos, exceto biológicos,
e dá outras providências.
Resultados de estudo de estabilidade acelerada (40°C ± 2°C / 75% UR ± 5% UR) do medicamento X 150 mg/comprimidos
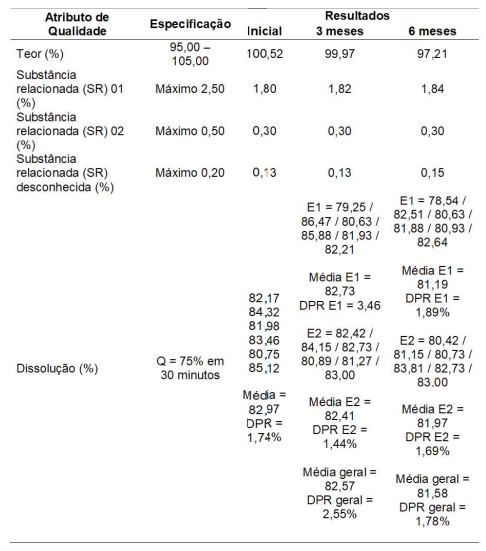
Sobre o estudo de estabilidade acima, avalie se são verdadeiras (V) ou falsas (F) as afirmações a seguir:
I – O medicamento X apresenta queda de teor no estudo de estabilidade acelerada. Por este motivo o estudo de estabilidade está reprovado.
II – É uma hipótese plausível que a SR 02 seja oriunda unicamente da síntese do IFA do medicamento.
III – É uma hipótese plausível que a SR 01 seja oriunda da degradação do fotolítica do IFA.
IV – Pelo conjunto dos resultados, pode-se concluir que o estudo de estabilidade acelerada atende aos requisitos para sua aprovação em conformidade com a zona climática II.
V – O estudo está aprovado frente aos requisitos da RDC 318/2019.
VI – Os resultados do ensaio de dissolução nas frequências de 3 e 6 meses estão reprovados em primeiro estágio.
De cima para baixo a ordem correta é:
Resultados de estudo de estabilidade acelerada (40°C ± 2°C / 75% UR ± 5% UR) do medicamento X 150 mg/comprimidos
Sobre o estudo de estabilidade acima, avalie se são verdadeiras (V) ou falsas (F) as afirmações a seguir:
I – O medicamento X apresenta queda de teor no estudo de estabilidade acelerada. Por este motivo o estudo de estabilidade está reprovado.
II – É uma hipótese plausível que a SR 02 seja oriunda unicamente da síntese do IFA do medicamento.
III – É uma hipótese plausível que a SR 01 seja oriunda da degradação do fotolítica do IFA.
IV – Pelo conjunto dos resultados, pode-se concluir que o estudo de estabilidade acelerada atende aos requisitos para sua aprovação em conformidade com a zona climática II.
V – O estudo está aprovado frente aos requisitos da RDC 318/2019.
VI – Os resultados do ensaio de dissolução nas frequências de 3 e 6 meses estão reprovados em primeiro estágio.
De cima para baixo a ordem correta é:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Análise, desenvolvimento e validação de metodologias para o controle físico-químico de produtos sujeitos à vigilância sanitária |
Q3341051
Farmácia
Uma das análises fundamentais na avaliação de ingredientes farmacêuticos é a avaliação de impurezas. Um
desafio especial é a determinação de substâncias relacionadas, visto que na maioria das vezes há uma relação
estrutural muito forte com o fármaco, tornando a separação
cromatográfica complexa. Um exemplo interessante é a
cetirizina (CTZ), antagonista H1 mostrada abaixo. A CTZ
tem sete relacionados principais, sendo mostrados abaixo
quatro relacionados, as substâncias de (A), (D), (F) e (G).
O cromatograma mostrado abaixo corresponde à injeção
de MR da CTZ e de todos os relacionados. As condições
cromatográficas específicas foram as do uso de coluna C18,
gradiente acetonitrila água, com detecção por arranjo de
diodos. A CTZ corresponde ao pico (4).:
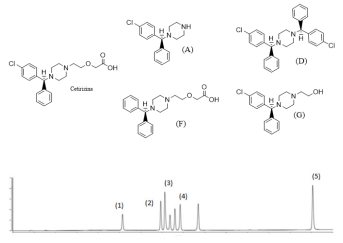
A correlação entre as substâncias (A), (D), (F) e (G) e os picos assinalados no cromatograma é:
A correlação entre as substâncias (A), (D), (F) e (G) e os picos assinalados no cromatograma é:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Análise, desenvolvimento e validação de metodologias para o controle físico-químico de produtos sujeitos à vigilância sanitária |
Q3341049
Farmácia
Para a realização da análise teor de produtos farmacêuticos por Clae é fundamental avaliar a adequação do
sistema analítico. É correto afirmar sobre a avaliação de
adequação de sistema, no caso específico das cinco substâncias apreendidas na farmácia de manipulação que:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Controle Microbiológico de Produtos sujeitos à Vigilância Sanitária |
Q3340661
Farmácia
Na análise microbiológica de controle da qualidade de
um xampu, são realizadas a determinação da carga microbiana e a pesquisa de patógenos. Assim, são verdadeiras
as afirmativas abaixo, EXCETO:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Controle Microbiológico de Produtos sujeitos à Vigilância Sanitária |
Q3340658
Farmácia
Em relação aos medicamentos e matérias primas não
estéreis:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Controle Microbiológico de Produtos sujeitos à Vigilância Sanitária |
Q3340657
Farmácia
Em relação à análise microbiológica de matérias primas e produtos não estéreis, assinale, entre as opções
abaixo, aquela que contempla a composição e forma de
apresentação dos produtos onde encontramos a MENOR
probabilidade de contaminação:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Controle Microbiológico de Produtos sujeitos à Vigilância Sanitária |
Q3340655
Farmácia
O conceito de “esterilidade” envolve a ausência de
todas as formas vivas de um produto, de acordo com a
metodologia empregada. De acordo com esse conceito,
pode-se dizer que:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Tecnologista em desenvolvimento de medicamentos |
Q3339676
Farmácia
Foi aberto um projeto de desenvolvimento de um medicamento contendo um antirretroviral da classe II do sistema
de classificação biofarmacêutica, fabricado como comprimidos de liberação imediata. Durante o desenvolvimento do
produto, foram avaliados os perfis de dissolução em 12 cubas nas condições descritas em monografia farmacopeica
sendo elas: 900 mL de meio tampão acetato pH 4,5 com
0,5% de lauril sulfato de sódio a 37 ºC, aparato cesta com
rotação de 75 rpm e com a quantificação por espectroscopia no ultravioleta-visível. Diferentes formulações foram
avaliadas e todas apresentaram semelhança no perfil de
dissolução, porém, foi observado um desvio padrão relativo
maior do que 20% nos primeiros tempos do perfil (5 e 10
minutos) e maior do que 10% nos demais tempos do perfil
(15, 30 e 45 minutos). Sobre o ensaio de perfil dissolução
em questão, pode-se afirmar que:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Tecnologista em desenvolvimento de medicamentos |
Q3339675
Farmácia
No desenvolvimento de um medicamento na forma
farmacêutica suspensão oral em veículo aquoso contendo
um insumo farmacêutico ativo (IFA) da classe II do sistema
de classificação biofarmacêutica, foram produzidos três
lotes experimentais sendo cada lote produzido com o IFA de
diferentes fabricantes. O Dossiê de Insumo Farmacêutico
Ativo (DIFA) dos três fabricantes apresenta a informação de
que o IFA é uma forma hidratada e apresenta informações
em relação à posição dos picos de difração em 2 teta. Estão
descritos no DIFA do fabricante A picos de difração em 5,5°,
7,2°, 12,8°, 15,7° e 21,4° com a análise feita com radiação
de cobre (λ =1,5406 Å), no do fabricante B em 5,5°, 7,3°,
12,7°, 15,8° e 21,4° com a análise feita com radiação de
cobre (λ =1,5406 Å) e no do fabricante C em 2,5°, 11,4°,
17,6°, 18,2°, 19,1° e 24,3° com a análise feita com radiação
de cobalto (λ=1,7890 Å). Considerando o exposto, observe
as afirmativas abaixo:
I – É plausível afirmar que o IFA do fabricante B apresenta a mesma forma sólida do fabricante A, não sendo possível garantir que ambos contêm uma única forma sólida, uma vez que os padrões de difração de raios X não foram apresentados.
II – Considerando a informação do DIFA que os IFAs são hidratos, os fabricantes B e C podem apresentar diferentes graus de hidratação ou são polimorfos do hidrato de mesma estequiometria.
III – A titulação Karl-Fisher não permite a diferenciação da água de superfície e da água e incorporada na estrutura cristalina do hidrato.
IV – Considerando a informação do DIFA que os IFAs são hidratos, os fabricantes A, B e C podem ser selecionados para o desenvolvimento do produto, pois, já que as propriedades físico-químicas dos IFAs são as mesmas não haverá diferenças no desempenho da formulação.
V – É importante a avaliação da interconversão entre as formas sólidas do IFA, uma vez que, em suspensão, pode ocorrer a transição para a forma estável, resultando em crescimento cristalino com consequente alteração na distribuição do tamanho das partículas, o que pode afetar a estabilidade física da suspensão.
Das afirmativas acima:
I – É plausível afirmar que o IFA do fabricante B apresenta a mesma forma sólida do fabricante A, não sendo possível garantir que ambos contêm uma única forma sólida, uma vez que os padrões de difração de raios X não foram apresentados.
II – Considerando a informação do DIFA que os IFAs são hidratos, os fabricantes B e C podem apresentar diferentes graus de hidratação ou são polimorfos do hidrato de mesma estequiometria.
III – A titulação Karl-Fisher não permite a diferenciação da água de superfície e da água e incorporada na estrutura cristalina do hidrato.
IV – Considerando a informação do DIFA que os IFAs são hidratos, os fabricantes A, B e C podem ser selecionados para o desenvolvimento do produto, pois, já que as propriedades físico-químicas dos IFAs são as mesmas não haverá diferenças no desempenho da formulação.
V – É importante a avaliação da interconversão entre as formas sólidas do IFA, uma vez que, em suspensão, pode ocorrer a transição para a forma estável, resultando em crescimento cristalino com consequente alteração na distribuição do tamanho das partículas, o que pode afetar a estabilidade física da suspensão.
Das afirmativas acima:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Tecnologista em desenvolvimento de medicamentos |
Q3339670
Farmácia
Em relação ao desenvolvimento de método de dissolução para formas farmacêuticas sólidas, avalie se são
verdadeiras (V) ou falsas (F) as afirmativas a seguir:
I – O perfil de dissolução é uma ferramenta importante durante o desenvolvimento do método, pois permite o estabelecimento das condições e das especificações mais adequadas para o controle do desempenho do produto.
II - Volumes de meio de dissolução que não atendam à condição sink podem ser utilizados desde que justificados e desde que haja comprovação da capacidade discriminativa do método.
III - A determinação do poder discriminativo do método de dissolução pode ser realizada por meio da inclusão de lotes que não foram capazes de demonstrar perfil farmacocinético aceitável in vivo.
As afirmativas I, II e III são respectivamente:
I – O perfil de dissolução é uma ferramenta importante durante o desenvolvimento do método, pois permite o estabelecimento das condições e das especificações mais adequadas para o controle do desempenho do produto.
II - Volumes de meio de dissolução que não atendam à condição sink podem ser utilizados desde que justificados e desde que haja comprovação da capacidade discriminativa do método.
III - A determinação do poder discriminativo do método de dissolução pode ser realizada por meio da inclusão de lotes que não foram capazes de demonstrar perfil farmacocinético aceitável in vivo.
As afirmativas I, II e III são respectivamente:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Tecnologista em desenvolvimento de medicamentos |
Q3339669
Farmácia
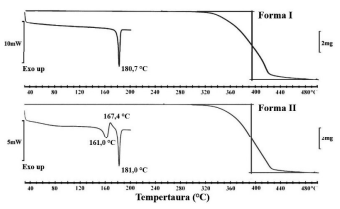
A figura abaixo apresenta as curvas de calorimetria diferencial exploratória (DSC) e de análise termogravimétrica
(TGA) de duas formas sólidas de um insumo farmacêutico
ativo (IFA). Sobre as figuras, pode-se afirmar que:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Tecnologista em desenvolvimento de medicamentos |
Q3339668
Farmácia
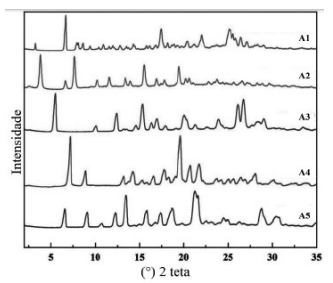
A figura abaixo apresenta os padrões experimentais de
difração de raios X de pó (DRXP) de 5 amostras (A1, A2, A3,
A4 e A5), obtidos nas mesmas condições de análise, de um
insumo farmacêutico ativo (IFA). A partir destes resultados,
pode-se interpretar que:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Tecnologista em desenvolvimento de medicamentos |
Q3339667
Farmácia
No estudo de degradação forçada, o insumo farmacêutico ativo e o produto acabado são expostos a condições de
estresse, como luz, temperatura, calor, umidade, hidrólise
ácida, hidrólise básica e oxidação. Sobre esse estudo
pode-se afirmar que:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Tecnologista em desenvolvimento de medicamentos |
Q3339666
Farmácia
Durante o processo de fabricação de comprimidos, é
possível observar alterações físicas no estado sólido do
fármaco. As transições de fase induzidas por processos
farmacêuticos devem se mostrar consistentes no estudo
de escalonamento para a escala produtiva quando um
protótipo é selecionado. Essas transições podem resultar
de diversas interações e condições durante as etapas de
produção, desde a mistura dos componentes até a obtenção
do comprimido final. Sobre alterações físicas induzidas por
processo, está correto afirmar que:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Tecnologista em desenvolvimento de medicamentos |
Q3339659
Farmácia
Considere as informações sobre a caracterização de
um insumo farmacêutico ativo e o seguinte contexto de
histórico de desenvolvimento:
(a) Insumo de classificação biofarmacêutica classe III.
(b) Perfil alvo de qualidade: cápsulas duras contendo 75 mg de IFA em 200 mg de peso médio teórico, com desintegração em até 10 minutos e perfil de dissolução muito rápido.
(c) Condições do ensaio de dissolução: meio 900 ml de tampão fosfato 6,8 a 37 1 °C, aparato pá com rotação de 50 rpm.
(d) Distribuição granulométrica do IFA: d10 – 2,2 micras; d50 – 36,5 micras; d90 – 252,8 micras
(e) Densidade livre e compactada do IFA: 0,44 g/ml e 0,32 g/ml, respectivamente
(f) Indice de Carr e Razão de Hausner do IFA: 27,3 e 1,37, respectivamente
(g) Ângulo de repouso do IFA: 60°
Observe as alternativas abaixo e avalie se são verdadeiras (V) ou falsas (F):
I - A estratégia de desenvolvimento deve considerar como primeira abordagem a mistura direta, pois os atributos de reologia do IFA são favoráveis a esta estratégia. Desta forma se assegura um produto de alta produtividade na planta fabril.
II - Por ser um IFA de classificação biofarmacêutica classe III, o ativo é um candidato a provas de bioisenção para demonstrar a sua eficácia e segurança.
III - Considerando que o IFA apresenta baixa solubilidade, trabalhar com especificações de distribuição granulométrica de valores reduzidos favorecerá os processos de dissolução a partir da forma farmacêutica.
IV - Considerando que o IFA apresenta alta solubilidade, é coerente explorar valores mais amplos de distribuição granulométrica e permitir a ampliação na especificação deste atributo de qualidade.
V - Uma vez confirmado que partículas com distribuição granulométrica mais ampla ainda atingem o perfil alvo de qualidade do medicamento, mantendo o desempenho de dissolução muito rápida, a especificação de granulometria pode ser classificada como um atributo não crítico de qualidade.
De cima para baixo, a ordem correta é:
(a) Insumo de classificação biofarmacêutica classe III.
(b) Perfil alvo de qualidade: cápsulas duras contendo 75 mg de IFA em 200 mg de peso médio teórico, com desintegração em até 10 minutos e perfil de dissolução muito rápido.
(c) Condições do ensaio de dissolução: meio 900 ml de tampão fosfato 6,8 a 37 1 °C, aparato pá com rotação de 50 rpm.
(d) Distribuição granulométrica do IFA: d10 – 2,2 micras; d50 – 36,5 micras; d90 – 252,8 micras
(e) Densidade livre e compactada do IFA: 0,44 g/ml e 0,32 g/ml, respectivamente
(f) Indice de Carr e Razão de Hausner do IFA: 27,3 e 1,37, respectivamente
(g) Ângulo de repouso do IFA: 60°
Observe as alternativas abaixo e avalie se são verdadeiras (V) ou falsas (F):
I - A estratégia de desenvolvimento deve considerar como primeira abordagem a mistura direta, pois os atributos de reologia do IFA são favoráveis a esta estratégia. Desta forma se assegura um produto de alta produtividade na planta fabril.
II - Por ser um IFA de classificação biofarmacêutica classe III, o ativo é um candidato a provas de bioisenção para demonstrar a sua eficácia e segurança.
III - Considerando que o IFA apresenta baixa solubilidade, trabalhar com especificações de distribuição granulométrica de valores reduzidos favorecerá os processos de dissolução a partir da forma farmacêutica.
IV - Considerando que o IFA apresenta alta solubilidade, é coerente explorar valores mais amplos de distribuição granulométrica e permitir a ampliação na especificação deste atributo de qualidade.
V - Uma vez confirmado que partículas com distribuição granulométrica mais ampla ainda atingem o perfil alvo de qualidade do medicamento, mantendo o desempenho de dissolução muito rápida, a especificação de granulometria pode ser classificada como um atributo não crítico de qualidade.
De cima para baixo, a ordem correta é:
Ano: 2024
Banca:
FIOCRUZ
Órgão:
FIOCRUZ
Prova:
FIOCRUZ - 2024 - FIOCRUZ - Tecnologista em Saúde Pública - Tecnologista em desenvolvimento de medicamentos |
Q3339656
Farmácia
O guia de estudo de estabilidade Nº 28 de 2019 apresenta recomendações para o cumprimento regulatório para
a realização dos estudos de estabilidade. Neste sentido,
avalie se são verdadeiras (V) ou falsas (F) as afirmativas
a seguir sobre os testes a serem realizados no estudo de
estabilidade, de acordo com o tipo de forma farmacêutica.
I – A quantificação de antioxidantes e conservantes, se estiverem presentes na formulação, é necessária para qualquer tipo de forma farmacêutica.
II – Se a pureza enantiomérica for importante para a eficácia e segurança do medicamento, deve ser comprovado que não ocorre racemização durante o estudo de estabilidade para qualquer tipo de forma farmacêutica.
III – A forma cristalina do insumo farmacêutico ativo no medicamento deve ser avaliada durante o estudo de estabilidade para qualquer forma farmacêutica, como comprimidos, cápsulas, soluções e suspensões.
IV – No caso de comprimidos dispersíveis, deve-se avaliar a dissolução, desintegração, umidade, dureza e friabilidade, sendo recomendada uma especificação de não mais que dez minutos para a desintegração.
As afirmativas I, II, III e IV são respectivamente:
I – A quantificação de antioxidantes e conservantes, se estiverem presentes na formulação, é necessária para qualquer tipo de forma farmacêutica.
II – Se a pureza enantiomérica for importante para a eficácia e segurança do medicamento, deve ser comprovado que não ocorre racemização durante o estudo de estabilidade para qualquer tipo de forma farmacêutica.
III – A forma cristalina do insumo farmacêutico ativo no medicamento deve ser avaliada durante o estudo de estabilidade para qualquer forma farmacêutica, como comprimidos, cápsulas, soluções e suspensões.
IV – No caso de comprimidos dispersíveis, deve-se avaliar a dissolução, desintegração, umidade, dureza e friabilidade, sendo recomendada uma especificação de não mais que dez minutos para a desintegração.
As afirmativas I, II, III e IV são respectivamente: