Foram encontradas 4.268 questões
Resolva questões gratuitamente!
Junte-se a mais de 4 milhões de concurseiros!
I – Sendo o registro do produto feito como medicamento genérico, o QTPP deve ser elaborado considerando a comparação da formulação entre o produto teste e o produto de referência, baseada principalmente nas características de desempenho do produto.
II – O QTPP pode incluir características do produto em questão, como uso pretendido, via de administração, dissolução, teor, assim como características de desempenho para os métodos analíticos utilizados, como precisão e exatidão.
III – O tempo de desintegração e o tempo de molhamento podem ser considerados no QTPP do produto, podendo incluir no perfil alvo qualquer característica que idealmente será alcançada para garantir a qualidade do medicamento.
IV – As características listadas no QTPP contribuem para a definição dos atributos críticos de qualidade (CQAs) que serão posteriormente identificados.
V – A elaboração do QTPP apresenta relação com o desenvolvimento de um medicamento com padrões de qualidade centrados no paciente.
Das afirmativas acima:
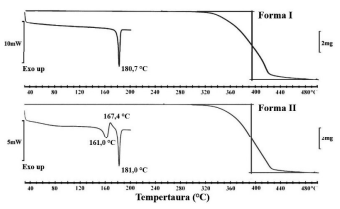
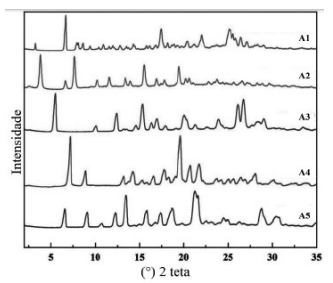
I – É plausível afirmar que o IFA do fabricante B apresenta a mesma forma sólida do fabricante A, não sendo possível garantir que ambos contêm uma única forma sólida, uma vez que os padrões de difração de raios X não foram apresentados.
II – Considerando a informação do DIFA que os IFAs são hidratos, os fabricantes B e C podem apresentar diferentes graus de hidratação ou são polimorfos do hidrato de mesma estequiometria.
III – A titulação Karl-Fisher não permite a diferenciação da água de superfície e da água e incorporada na estrutura cristalina do hidrato.
IV – Considerando a informação do DIFA que os IFAs são hidratos, os fabricantes A, B e C podem ser selecionados para o desenvolvimento do produto, pois, já que as propriedades físico-químicas dos IFAs são as mesmas não haverá diferenças no desempenho da formulação.
V – É importante a avaliação da interconversão entre as formas sólidas do IFA, uma vez que, em suspensão, pode ocorrer a transição para a forma estável, resultando em crescimento cristalino com consequente alteração na distribuição do tamanho das partículas, o que pode afetar a estabilidade física da suspensão.
Das afirmativas acima:
Coluna I
1. abordagem mínima. 2. abordagem aprimorada.
Coluna II
( ) principalmente empírica com o desenvolvimento geralmente realizado avaliando-se uma variável por vez.
( ) a qualidade do medicamento é garantida por uma estratégia de controle baseada em riscos.
( ) o gerenciamento do ciclo de vida é reativo (ou seja, solução de problemas e ação corretiva).
( ) o processo de fabricação é ajustável dentro do design space.
( ) abordagem de ciclo de vida para validação de processo, idealmente com a verificação continuada do processo.
A sequência correta, de cima para baixo, é:
I – É permitido que a empresa solicite o registro de um medicamento em mais de um material de embalagem primária.
II – O vidro tipo I, também denominado vidro borossilicato, é o único tipo de vidro que pode ser utilizado para medicamentos de uso humano por ser totalmente inerte e não trazer riscos de incompatibilidade com a forma farmacêutica.
III – Polímeros termoplásticos podem ser usados para a tecnologia blow-fill-seal, na qual as etapas de formação da embalagem, enchimento com o produto farmacêutico e selagem do sistema final ocorrem em operação contínua.
IV – Embalagens termomoldáveis devem conter plastificantes em suas composições, permitindo a redução da temperatura de transição vítrea dos polímeros para que as temperaturas de processo possam ser reduzidas. São exemplos de plastificantes de embalagens termomoldáveis os ftalatos.
As afirmativas I, II e III e IV são respectivamente:
I – O perfil de dissolução é uma ferramenta importante durante o desenvolvimento do método, pois permite o estabelecimento das condições e das especificações mais adequadas para o controle do desempenho do produto.
II - Volumes de meio de dissolução que não atendam à condição sink podem ser utilizados desde que justificados e desde que haja comprovação da capacidade discriminativa do método.
III - A determinação do poder discriminativo do método de dissolução pode ser realizada por meio da inclusão de lotes que não foram capazes de demonstrar perfil farmacocinético aceitável in vivo.
As afirmativas I, II e III são respectivamente:
A embalagem farmacêutica tem as funções de conferir contenção, proteção, identificação, informação e conveniência para promover a adesão do paciente ao tratamento farmacêutico. Sobre as embalagens farmacêuticas, avalie se são verdadeiras(V) ou falsas(F) as afirmativas a seguir:
I – A integridade de uma embalagem farmacêutica é fundamental para garantir a manutenção de requisitos essenciais que assegurem a eficácia, segurança, uniformidade de conteúdo, pureza, estabilidade química, física e microbiológica do medicamento.
II – Embalagens de alta barreira para vapores são indicadas para produtos que degradam por exposição à luz ultravioleta.
II – Frascos de policloreto de vinila (PVC) lacrados com tampa com sílica são equivalentes em barreira de proteção de umidade quando comparados aos frascos de vidro.
De cima para baixo, a ordem correta é:
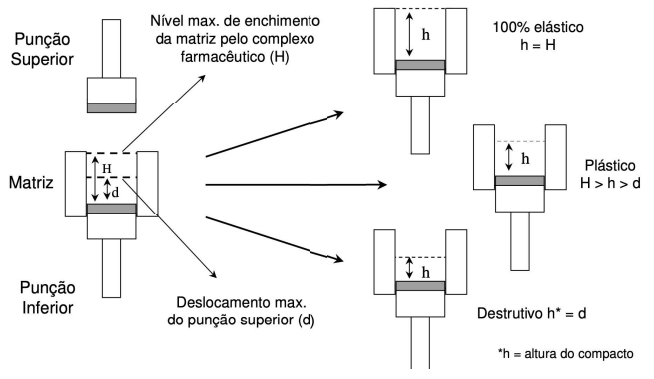
Representação das deformações do tipo elástica e plástica. Onde h = altura do comprimido.